RAD
Retrieval Augmented Docking
Connect to our public HNSW service with your own scoring function and begin virtual screening of 2 billion molecules in minutes
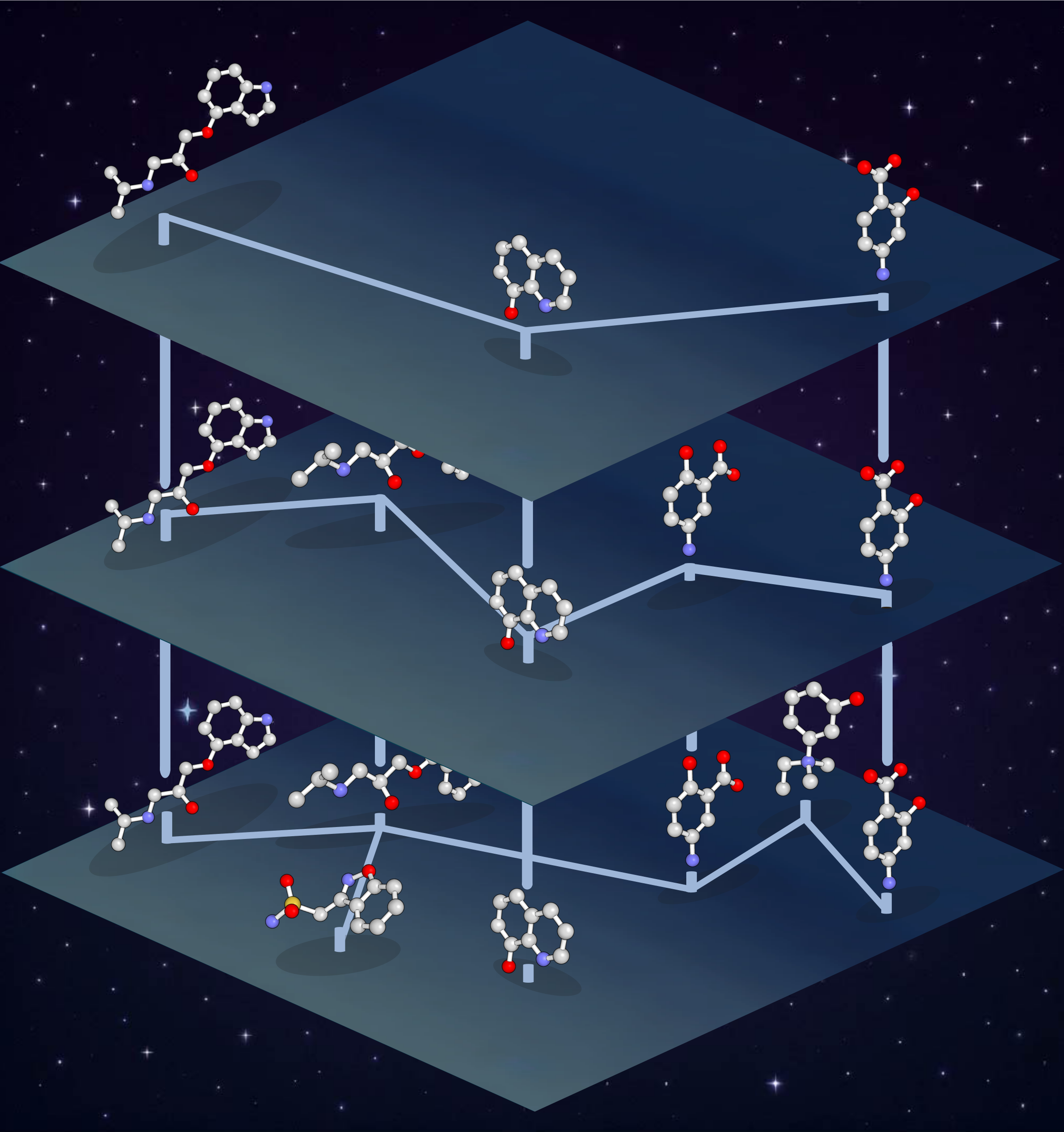
Sparse hierarchy allows rapid identification of promising areas of chemical space
Graph edges are based on Tanimoto similarity and chosen to maintain the small-world property, enabling efficient exploration
User-defined scoring functions guide graph traversal to quickly find good-scoring molecules while avoiding unpromising areas of chemical space
Lightweight scoring workers coordinate with a single central process to enable massively parallel scoring
RAD can find over 50% of the top-scoring molecules in a library, while doing the expensive scoring calculation for only 1%.
Proven performance on billion-scale chemical libraries with traditional molecular docking and modern ML models.
Scale from single machines to HPC clusters with Redis-coordinated workers that can run anywhere.
Deploy locally, use our hosted public service to avoid setup complexity, or run your own infrastructure.
Read the original paper describing the method and showcasing the performance on the DUDE-Z dataset.
Read PaperSee RAD applied to billion-scale chemical libraries with UCSF DOCK and the ML model Chemprop.
Read Paper